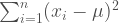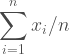In high school people are usually taught the three trigonometric functions, which is odd because there are a lot more than that, nine that see regular use. They can be thought of as three pairs of three starting with the sine, cosine, and tangent. It takes a bit of work to draw each of these functions so the code is presented at the end of the post rather than with each graphic.
Let’s draw a circle and use it to construct the values of the functions. All of them correspond to a characteristic of a triangle draw on the circle. Let’s start with the sine and cosine because they have the most natural representations.
Here are the sine and cosine:
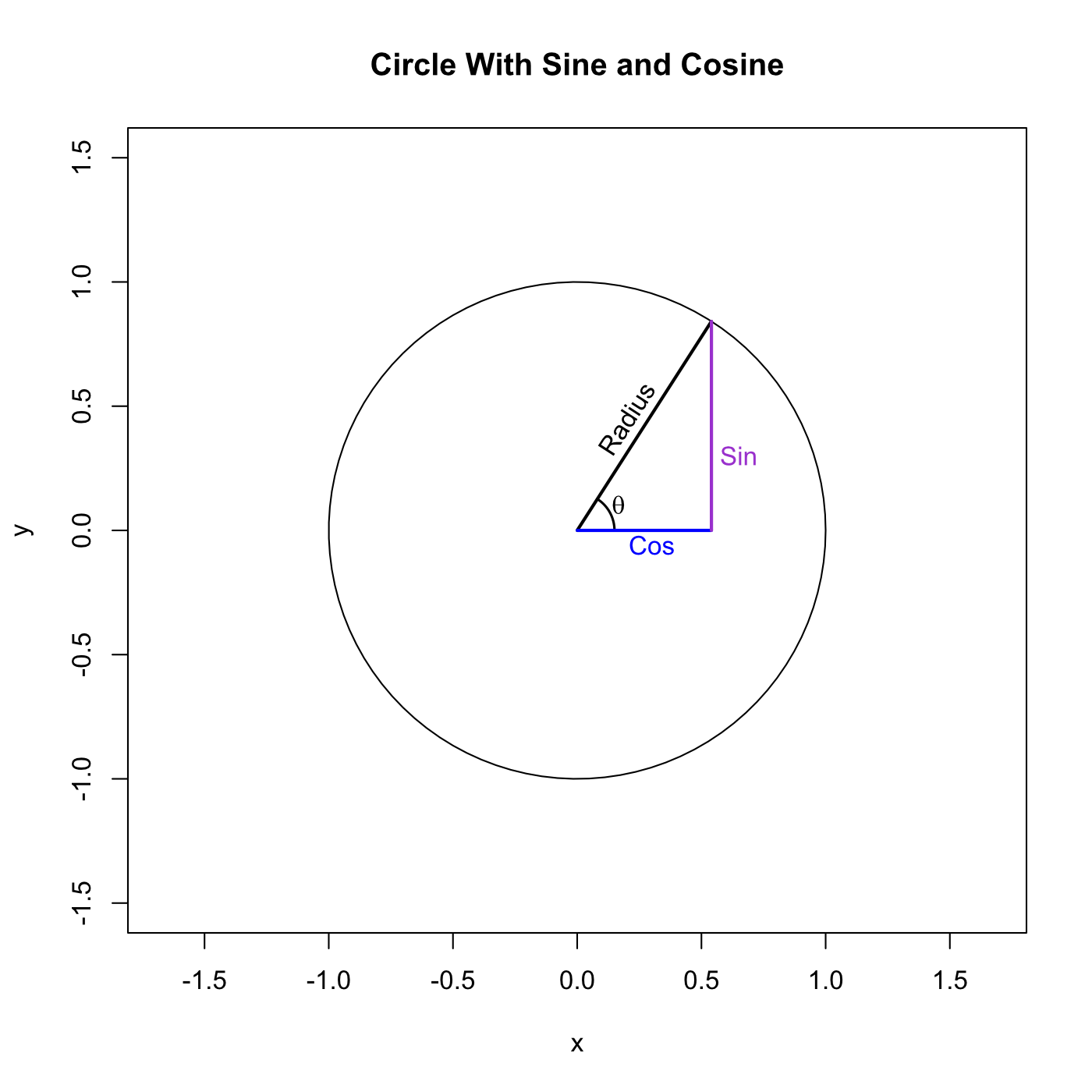
They have the length of those two colored lines respectively. The length of those lines is completely determined by the length of the radius and the angle of theta. In this case the angle is 1 radian and the radius of the unit circle is exactly equal to 1 so we can use R to find that . . .
sin(1) [1] 0.841471 cos(1) [1] 0.5403023
In other words the sine of theta gives us the y-coordinate of a point on the unit circle and the cosine of theta gives us the x-coordinate of that same point. Just by looking at the image we can intuit certain properties of the two functions. Both of them vary between -1 and 1, the bounds of the circle, and they’re offset by exactly 90 degrees (π/2 radians) which means that the sine of π is the same the cosine of π/2.
If we plot how the vales of the sin and cosine change with in inputs we get a pair of waves.
x <- seq(-5,5,.1) y1 <- sin(x) y2 <- cos(x) plot(y1~x, main = 'Sine and Cosine Waves', lwd = 3, col = 'blue', type = 'l', ylim = c(-2,2)) lines(y2~x, lwd = 3, col = 'darkorchid')
These are are the shape that you see on oscilloscopes in old science fiction movies (or at work if you often measure voltages). You can see that there are points where the two curves cross at all of the multiples of π/4 (a 45 degree angle).
Notice that the three lines (radius, sine, and cosine) together describe a right triangle. Because of this the trigonometric functions can be used to solve triangles and then to solve much more complex structures because we can build them out of triangles. If we vary the radius of the circle the length of the other lines scale proportionally. In fact this is true for all of the trigonometric functions. As a result we can simply a lot of things by working on the unit circle and then scaling appropriately when we’re done.
Let’s use this fact to derive an important fact about the sine and cosine.
Using the sine, cosine, and tangent it is possible to fully describe a right triangle with just a few pieces of information. Consider a simple right triangle on a circle with angles of A = 1, B = 0.57, the third angle is π/2 since its a right angle.
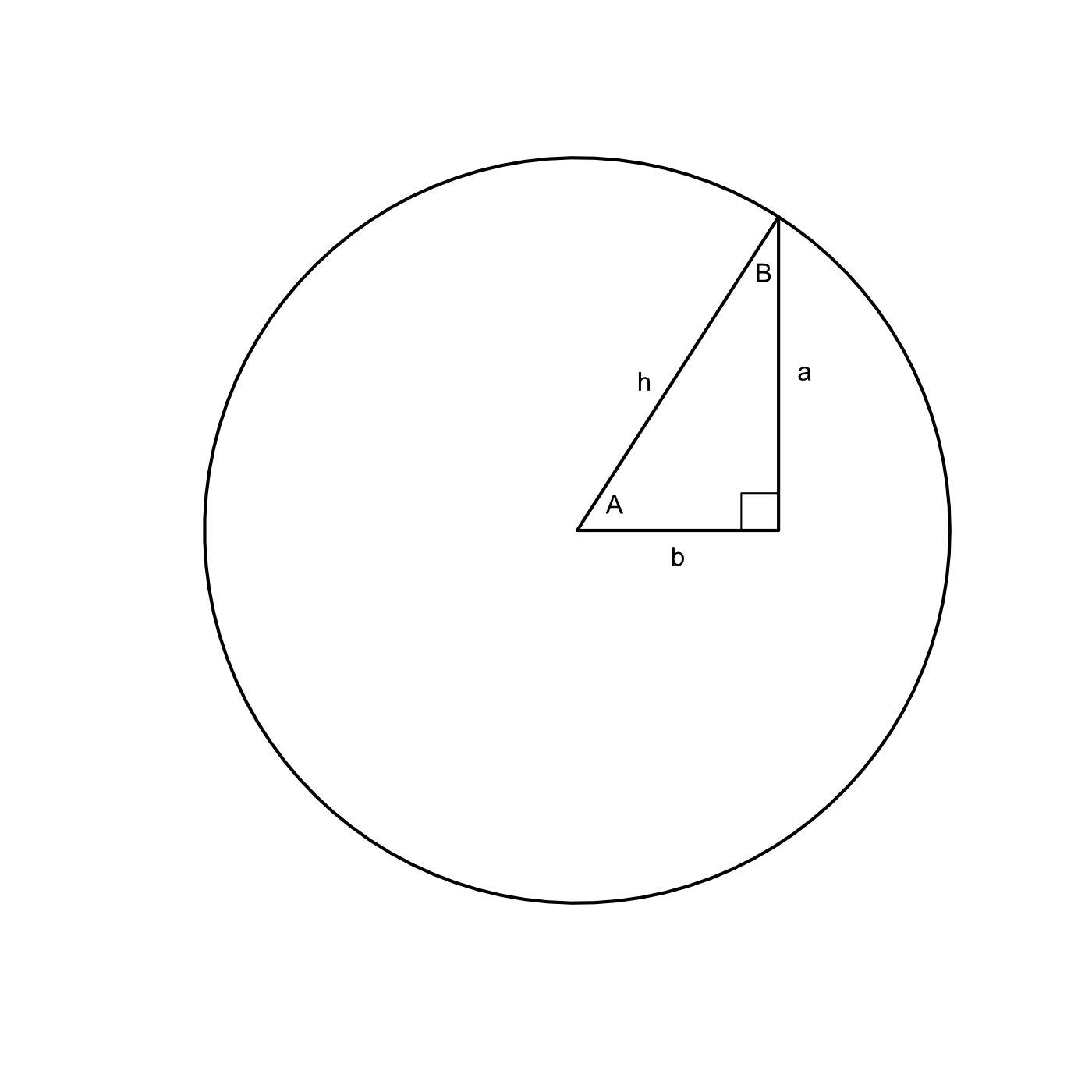
The sine of angle A is 0.84 so we know that side a has a length of 0.84r and the cosine of A is 0.54 so we know that the length of side b is 0.54r, where r is the length of the hypotenuse (or the radius). This follows quite obviously from the definition of sine and cosine and the properties we’ve discussed. The only piece of information we don’t have is the length of the radius since we don’t know if this is a unit circle or not.
Can we calculate it?
Actually no, we cannot. There are an infinite number of valid triangles with those angles!
That’s unfortunate but considering what would happen if we knew that the length of side a is very illustrative. If the hypotenuse was exactly equal to 2 we knew that we could solve the entire triangle! Why? Consider again the value which we calculated which was 0.84r. That 0.84 is constant, only the r can vary as the size of the triangle increases or decreases.
In other words . . .
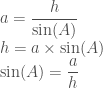
So if the angle A 1 and the length of the corresponding side is 2. . .
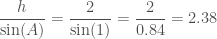
This is where the identities taught to school children as “sine equals opposite over hypotenuse” and “cosine equals adjacent over hypotenuse” come from. Intuitively if the length of the sides is determined by the hypotenuse and the angle then the angle must be determined by the hypotenuse and the sides!
Next up is the tangent function!
Code for drawing the sine and cosine. The circ() function is not from base R, we created last time.
circ(xlim = c(-1.5,1.5), ylim = c(-1.5,1.5), lwd = 1, main = 'Unit Circle With Sine and Cosine') segments(0, 0, cos(1), sin(1), lwd = 2) text(.20, .45, 'Radius', srt = 57) lines(th, lwd = 1.5) text(.17, .1, expression(theta)) segments(0, 0, cos(1), 0, lwd = 2, col = 'blue') text(.3, -.05, 'Cos', col = 'blue') segments(cos(1), sin(1), cos(1), 0, lwd = 2,col = 'darkorchid') text(.65, .3, 'Sin', col = 'darkorchid')
Drawing the triangle to solve. Basically the same shape with different labels.
segments(0, 0, cos(1), sin(1), lwd = 2) segments(cos(1), sin(1), cos(1), 0, lwd = 2) segments(0, 0, cos(1), 0, lwd = 2) text(.1, 0.07, 'A') text(cos(1)+.07, sin(1)/2, 'a') text(cos(1)/2, -0.07, 'b') text(cos(1)-.04, sin(1)-.15, 'B') text(cos(1)/3, .4, 'h') rect(cos(1)-.1,0,cos(1),.1)